 Minotauromaquia
Minotauromaquia
Bei der Vermarktung eines Medizinprodukts benötigen Sie eine Freigabe der Food and Drug Administration (FDA). Es ist ein strenger Prozess und es gibt erhebliche Gebühren. Das medizinische Gerät fällt unter 3 Klassen und die erforderlichen Einreichungen hängen davon ab, welcher Klasse Ihr Gerät zugewiesen ist. Informationen, die für den FDA-Zulassungsprozess relevant sind, sind in die folgenden Abschnitte unterteilt, so dass Sie die Schritte leicht nachvollziehen können.
Schritte
-
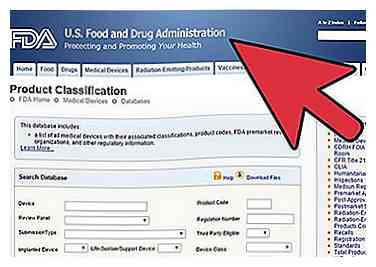 1 Ermitteln Sie die Klassifizierung Ihres Medizinproduktes.
1 Ermitteln Sie die Klassifizierung Ihres Medizinproduktes. - Es gibt 3 Klassen: Klasse I, II und III. Sie werden nach Risiko klassifiziert, wobei die Klasse I die niedrigste und die Klasse III die größte ist.
- Sie erfahren anhand der Klassifizierung Ihres Geräts, welche Prä-Marketing-Einreichung von der FDA gefordert wird. Allgemeine Kontrollen werden als Klasse I entweder mit oder ohne Ausnahmen zugewiesen. Allgemeine Kontrollen und besondere Kontrollen werden als Klasse II entweder mit oder ohne Ausnahmen zugewiesen. Allgemeine Kontrollen und die Zulassung vor dem Inverkehrbringen werden als Klasse III zugewiesen.
- Jedes Gerät hat eine Regulierungsnummer. Sie benötigen diese Nummer, um die Klassifizierung Ihres Geräts zu bestimmen. Gehen Sie zur Datenbank auf der Website http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpcd/classification.cfm.
- Suchen Sie nach einem Teil des Namens Ihres medizinischen Geräts oder, wenn Sie das medizinische Fachgebiet kennen, geben Sie es dort ein, wo es Geräte-Panel heißt.
-
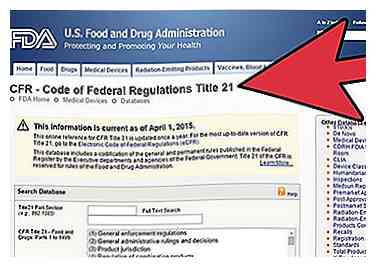 2 Suchen Sie nach der Regulierung für Ihr Gerät, nachdem Sie das Gerätepanel zu CFR oder Code of Federal Regulation Title 21 gefunden haben.
2 Suchen Sie nach der Regulierung für Ihr Gerät, nachdem Sie das Gerätepanel zu CFR oder Code of Federal Regulation Title 21 gefunden haben. - Gehen Sie zur CFR-Suchseite unter http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfCFR/CFRSearch.cfm.
- Lesen Sie die Informationen zu Vorschriften wie allgemeine Bestimmungen, Verpackung und Kennzeichnung und andere relevante Informationen zu CFR.
-
 3 Finden Sie heraus, ob Sie FDA-Zulassung erhalten können, um Ihr Medizinprodukt zu vermarkten.
3 Finden Sie heraus, ob Sie FDA-Zulassung erhalten können, um Ihr Medizinprodukt zu vermarkten. - Wenn Ihr Gerät ohne Ausnahmen unter Klasse I oder II fällt, benötigen Sie eine Vorabbenachrichtigung oder 510K für die Vermarktung.
- Wenn Ihr Gerät unter die Klasse III fällt, benötigen Sie eine Zulassung vor dem Inverkehrbringen oder PMA, es sei denn, es handelt sich um ein Gerät vor der Änderung (das heißt auf dem Markt vor der Änderung der Medizinprodukte 1976 oder ein gleichwertiges Gerät). In diesem Fall benötigen Sie eine 510K.
-
 4 Reichen Sie Ihre Pre-Market Notification oder 510K bei Bedarf ein.
4 Reichen Sie Ihre Pre-Market Notification oder 510K bei Bedarf ein. - Eine Liste der 510k-Formulare finden Sie unter http://www.fda.gov/MedicalDevices/. Geben Sie im Such-Tool das Stichwort "Pre-market Notification Form" ein. Wählen Sie den Link zu 510k Review Gebühren und Sie haben die Möglichkeit, aus dem Menü alle notwendigen Informationen für 510k Einreichungsprozess auszuwählen.
-
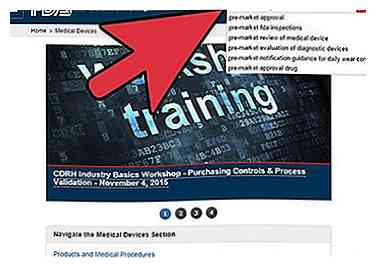 5 Reichen Sie Ihre Anwendung für die Marktzulassung oder PMA ein, falls zutreffend.
5 Reichen Sie Ihre Anwendung für die Marktzulassung oder PMA ein, falls zutreffend. - Gehen Sie zu http://www.fda.gov/MedicalDevices/ und geben Sie im Suchwerkzeug das Stichwort "Pre-Market Approval" ein. Wählen Sie den Link zur Pre-Market Approval (PMA) und Sie werden auf die Seite aufgefordert, auf der Sie Ihre Bewerbung einreichen und alle notwendigen Informationen für den PMA-Prozess erhalten können.
 1 Ermitteln Sie die Klassifizierung Ihres Medizinproduktes.
1 Ermitteln Sie die Klassifizierung Ihres Medizinproduktes.  2 Suchen Sie nach der Regulierung für Ihr Gerät, nachdem Sie das Gerätepanel zu CFR oder Code of Federal Regulation Title 21 gefunden haben.
2 Suchen Sie nach der Regulierung für Ihr Gerät, nachdem Sie das Gerätepanel zu CFR oder Code of Federal Regulation Title 21 gefunden haben.  3 Finden Sie heraus, ob Sie FDA-Zulassung erhalten können, um Ihr Medizinprodukt zu vermarkten.
3 Finden Sie heraus, ob Sie FDA-Zulassung erhalten können, um Ihr Medizinprodukt zu vermarkten.  4 Reichen Sie Ihre Pre-Market Notification oder 510K bei Bedarf ein.
4 Reichen Sie Ihre Pre-Market Notification oder 510K bei Bedarf ein.  5 Reichen Sie Ihre Anwendung für die Marktzulassung oder PMA ein, falls zutreffend.
5 Reichen Sie Ihre Anwendung für die Marktzulassung oder PMA ein, falls zutreffend. Facebook
Twitter
Google+